Look Beyond the Brain: A Case of Insulinomatosis Disguised as Frontal Lobe Epilepsy and Review of the Literature
Introduction
Pancreatic neuroendocrine tumors (PanNETs) are uncommon neoplasms that account for fewer than 2% of all pancreatic malignancies,with an estimated annual incidence of approximately 1 per 100,000 individuals in Western populations [1,2]. Among functional PanNETs, insulinomas are the most prevalent subtype, occurring at an incidence of approximately 1–4 per million person-years [2,3]. The vast majority of insulinomas are solitary, benign, and surgically curable; however, up to 10% are multiple, malignant, or associated with hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN1) [3,4]. In rare instances, patients present with a distinct multicentric pattern of insulin-secreting tumors that cannot be attributed to MEN1 or other recognized genetic syndromes, a condition now classified as insulinomatosis. colleagues in 2009, who characterized 14 patients with multicentric insulinoma disease that was distinguished from both sporadic solitary insulinoma and MEN1- associated tumors [5] . The condition is defined by the synchronous or metachronous occurrence of multiple insulin-expressing PanNETs of varying sizes, accompanied by diffuse insulin-expressing monohormonal endocrine cell clusters (IMECCs) and neuroendocrine microadenomatosis arising from widespread β-cell proliferation throughout the pancreatic parenchyma [5,6]. This diffuse tumorigenic process sets insulinomatosis apart from solitary sporadic insulinoma, in which a single well-circumscribed tumor can typically be excised with negligible risk of recurrence, and from MEN1-associated multicentric tumors, which occur within the broader context of germline MEN1 mutations and multi-organ endocrinopathies [5,7] .
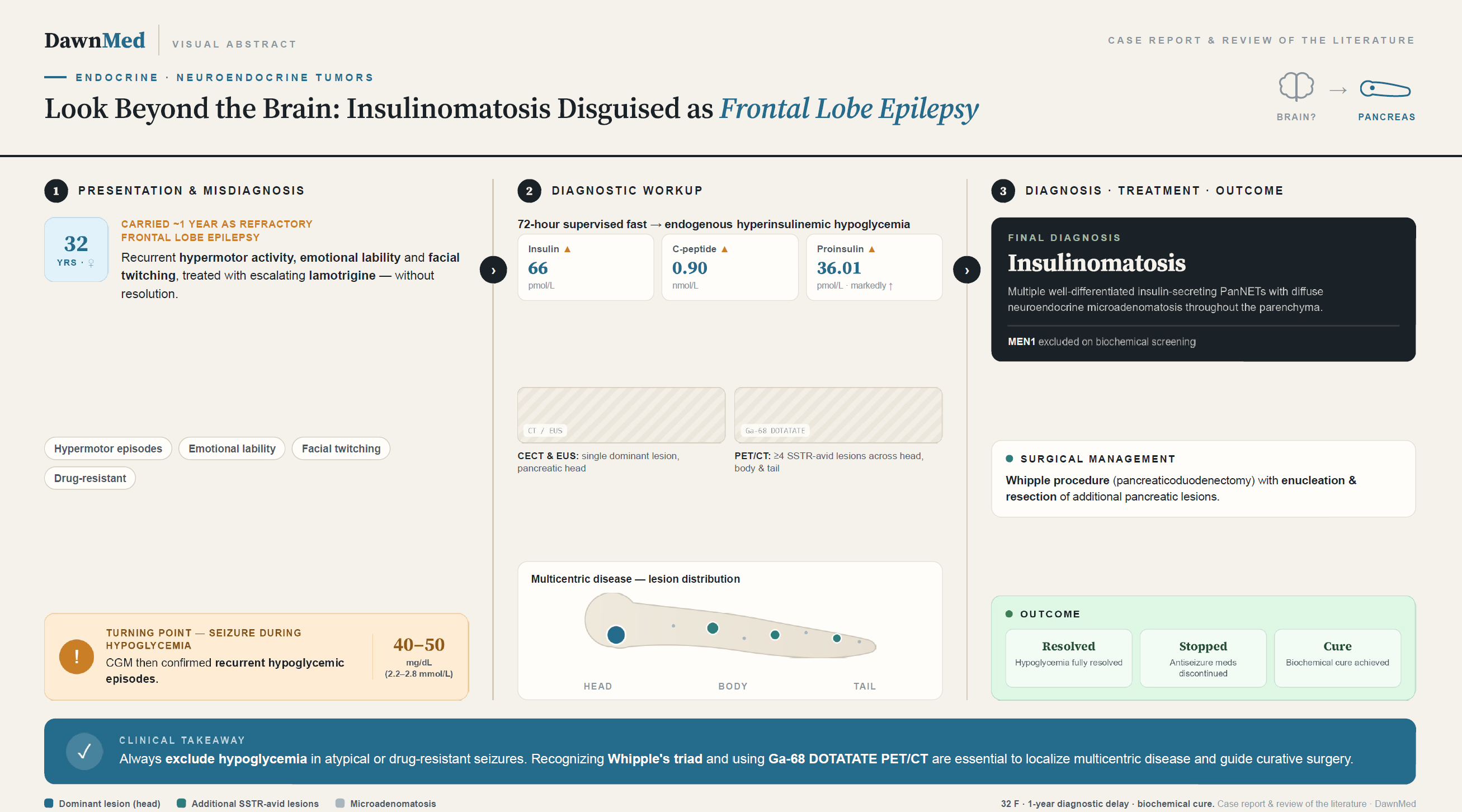
Clinically, insulinomatosis presents with the hallmark features of endogenous hyperinsulinemic hypoglycemia (EHH), including Whipple’s triad: symptoms consistent with hypoglycemia, a documented low plasma glucose concentration at the time of symptoms, and resolution of symptoms upon correction of hypoglycemia [8]. Neuroglycopenic manifestations including confusion, behavioral disturbance, seizures, and loss of consciousness dominate the clinical presentation and frequently lead to initial misdiagnosis as a primary neurological or psychiatric disorder [9,10]. In a landmark series of 59 insulinoma patients, neurological disorders constituted the initial misdiagnosis in 64% of cases, with epilepsy specifically diagnosed in 39% [9]. The diagnostic delay from symptom onset to definitive diagnosis of insulinoma averages 1.5 to 3 years in most series, with some patients enduring misdiagnosis for decades [9]. This delay is compounded in insulinomatosis by the multifocal nature of the disease, which may not be fully appreciated by conventional anatomic imaging modalities.
Histopathologically, insulinomatosis is characterized by the coexistence of macroscopic insulin-producing tumors (typically well-differentiated, low-grade PanNETs) and microscopic precursor lesions, including microadenomas (<5 mm) and IMECCs (<1 mm), both demonstrating insulin immunoreactivity [5,6]. These proliferative microlesions are considered to represent the biological substrate for the high recurrence rate observed in insulinomatosis: in the original Anlauf series, recurrent hypoglycemia developed in 43% of patients, with a mean time to relapse of 8.4 years [5]. More recent literature reports an even higher recurrence burden, with all sporadic cases in one series experiencing at least one relapse and 41% requiring two or more surgical resections [11] . The molecular underpinnings of insulinomatosis remain incompletely elucidated; however, germline mutations in the MAFA gene, encoding a β-cell–enriched transcription factor, have been identified as the cause of familial insulinomatosis in two unrelated kindreds [12,13]. The majority of sporadic cases, however, lack identifiable MAFA mutations, suggesting alternative pathogenic mechanisms yet to be characterized [12]
Advanced functional imaging, particularly Ga-68 DOTATATE positron emission tomography/ computed tomography (PET/CT), has significantly improved the detection of multifocal PanNETs, with reported sensitivities of 87.5–90% for insulinoma localization, considerably surpassing conventional CT (55%) and MRI (61%) [14,15]. The integration of functional imaging into the diagnostic algorithm has proven critical for accurate surgical planning in insulinomatosis, where the extent of disease dictates the choice between parenchyma-sparing enucleation and more extensive resection such as Pancreaticoduodenectomy or subtotal pancreatectomy [5,11] .
Despite growing recognition, insulinomatosis remains a significantly underdiagnosed entity, with approximately 108 cases reported in the literature to date [11,16]. Its rarity, combined with the clinical overlap between neuroglycopenic hypoglycemia and primary seizure disorders, creates a formidable diagnostic challenge that frequently results in prolonged misdiagnosis and inappropriate treatment. We report the case of a 32-year-old woman who carried a diagnosis of refractory frontal lobe epilepsy for one year before the identification of insulinomatosis as the underlying cause of her symptoms. This case illustrates the diagnostic pitfalls inherent in the evaluation of drug-resistant seizures, highlights the indispensable role of functional imaging in delineating multifocal pancreatic disease, and underscores the principles of comprehensive surgical management required for achieving biochemical cure.
Case Presentation
A 32-year-old woman with a known history of frontal lobe epilepsy and chronic inactive hepatitis B virus (HBV) infection presented to the emergency department with recurrent seizure attacks associated with documented hypoglycemia.
The patient had been diagnosed with epilepsy approximately one year prior to presentation and was under regular neurological follow-up. Her episodes were characterized by dilated pupils, changes in voice tone, rapid speech, inappropriate laughing or crying, hypermotor activity with facial twitching and numbness, sometimes followed by running, sleep, and normal recovery with full recall of events. Electroencephalography (EEG) performed during episodes demonstrated frontal lobe epileptiform activity, while interictal EEG, brain magnetic resonance imaging (MRI), and single-photon emission computed tomography (SPECT) were unremarkable. She was initiated on lamotrigine with subsequent dose escalation due to persistent seizure activity.
Despite antiseizure therapy, episodes continued. Several weeks before the current presentation, she was hospitalized following a seizure associated with documented hypoglycemia. She subsequently began continuous glucose monitoring (CGM), which revealed multiple hypoglycemic episodes with glucose values reaching 40–50 mg/dL (2.2–2.8 mmol/L). A comprehensive review of systems was otherwise unremarkable.
Past History, Medications, and Social History
Her past medical history was significant for frontal lobe epilepsy and chronic inactive HBV infection. Past surgical history was unremarkable. She reported an allergy to contrast media. Appropriate pre-medication with corticosteroids and antihistamines was administered prior to all contrast-enhanced imaging studies, and no adverse reactions occurred. Her only medication was lamotrigine 150 mg twice daily. There was no family history of seizures, endocrine disorders, or liver disease. She denied smoking, alcohol intake, recreational drug use, or use of herbal or hepatotoxic supplements.
Physical examination
On examination, the patient was vitally stable (weight 78.9 kg, height 169 cm, BMI 27.6 kg/m²). Physical examination, including a detailed neurological assessment, was unremarkable with no focal deficits. Thyroid examination was normal.
Investigations
Initial laboratory investigations, including complete blood count, renal function, and liver function tests, were within normal limits. Serial capillary and venous glucose measurements confirmed recurrent hypoglycemia, with glucose values as low as 1.9 mmol/L. The lowest recorded value of 1.9 mmol/L was obtained via venous sampling while the patient was symptomatic (tremulousness and confusion), thereby satisfying Whipple’s triad. Hemoglobin A1c was 4.9%, consistent with recurrent hypoglycemic episodes (Table 1).
Table 1. Selected Glucose Monitoring Results
Parameter | Patient Value | Reference Range |
Glucose (random) | 1.9 mmol/L | 3.9–11.1 mmol/L |
Glucose (fasting) | 2.20 mmol/L | 3.9–7.8 mmol/L |
HbA1c | 4.9% | 4.0–5.6% |
A supervised 72-hour fasting test confirmed endogenous hyperinsulinemic hypoglycemia, demonstrating inappropriately elevated insulin and C-peptide levels in the setting of hypoglycemia, along with markedly elevated proinsulin. Insulin antibody testing and oral antidiabetic drug (sulfonylurea) screening were both negative, excluding exogenous or pharmacologic causes of hypoglycemia (Table 2).
Table 2. 72-Hour Fasting Test Results
Parameter | Patient Value | Reference Range |
Plasma Glucose | 2.6 mmol/L | 3.9–5.5 mmol/L |
Insulin | 66 pmol/L | < 18 pmol/L |
C-Peptide | 0.90 nmol/L | 0.3–1.3 nmol/L |
Proinsulin | 36.01 pmol/L | < 7.25 pmol/L |
Insulin Antibodies (IAA) | < 0.40 U/mL | < 0.40 U/mL |
Oral Antidiabetic Drug Screen | Negative | — |
A glucagon challenge test was performed, with the first measurements obtained at 10 minutes postinjection (no pre-glucagon baseline was recorded), which demonstrated a brisk glycemic response following glucagon administration with persistently elevated insulin levels, further supporting autonomous insulin secretion (Table 3a). A short Synacthen (cosyntropin) stimulation test confirmed adequate adrenal reserve (baseline cortisol 501.2 nmol/L, peak 670.1 nmol/L at 30 minutes), thereby excluding adrenal insufficiency as a contributing cause of hypoglycemia (Table 3b).
Table 3a. Glucagon Challenge Test
Time (minutes) | Blood Glucose (mmol/L) | Insulin (pmol/L) | C-Peptide (nmol/L) |
10 | 3.8 | 66.34 | 0.90 |
20 | 6.5 | 78.15 | 0.82 |
30 | 6.2 | 64.47 | 0.65 |
Table 3b. Short Synacthen (Cosyntropin) Stimulation Test
Parameter | Value |
ACTH (baseline) | 21.74 pmol/L |
Cortisol (baseline) | 501.2 nmol/L |
Cortisol (30 minutes) | 670.1 nmol/L |
Cortisol (60 minutes) | 617.2 nmol/L |
Additional endocrine investigations, including IGF-1 (205 and 212 ng/mL), FSH (2.53 IU/L), and TSH (1.91 mIU/L), were within normal limits.
Imaging
Contrast-enhanced computed tomography (CECT) of the abdomen and pelvis revealed a heterogeneously enhancing space-occupying lesion in the pancreatic head with a hypodense non-enhancing area suggestive of necrosis, along with a few peripancreatic lymph nodes. The lesion closely abutted the superior mesenteric vein (SMV) without definite encasement or invasion, raising suspicion for an atypical insulinoma.
Endoscopic ultrasound (EUS) confirmed a welldemarcated, 30 × 20 mm, round, slightly hypoechoic lesion in the pancreatic head with two small areas of possible cystic degeneration. The mass was close to the SMV, appearing to touch it but without evidence of invasion. The common bile duct (CBD) was slender and the main pancreatic duct (MPD) was not dilated. Color Doppler was applied, and two passes with a 22G Acquire fine-needle biopsy (FNB) needle yielded adequate tissue for histological examination.
Histopathological examination of the EUS-guided biopsy confirmed a well-differentiated pancreatic neuroendocrine tumor (PanNET), Grade 1 (low grade).
Given the confirmed neuroendocrine tumor, biochemical screening for multiple endocrine neoplasia type 1 (MEN1) was performed. Prolactin and chromogranin A levels were within normal limits. Parathyroid hormone (PTH) was elevated at 14.75 pmol/L in the setting of low 25-hydroxyvitamin D (19.9 nmol/L), normal adjusted calcium, and lownormal phosphorus—findings most consistent withsecondary hyperparathyroidism due to vitamin D deficiency rather than primary hyperparathyroidism (Table 4). MEN1 genetic testing was not performed during the index admission; however, it has been planned for the upcoming follow-up visit.
Table 4. MEN1 Screening Panel
Parameter | Patient Value | Reference Range |
Prolactin | 467.54 mIU/L | 102–496 mIU/L |
PTH | 14.75 pmol/L | 1.6–6.9 pmol/L |
25-OH Vitamin D | 19.9 nmol/L | 50–125 nmol/L |
Phosphorus | 0.77 mmol/L | 0.8–1.5 mmol/L |
Adjusted Calcium | 2.3 mmol/L | 2.2–2.6 mmol/L |
Magnesium | 0.69 mmol/L | 0.7–1.0 mmol/L |
Chromogranin A | 60 ng/mL | < 108 ng/mL |
Ga-68 DOTATATE positron emission tomography/computed tomography (PET/CT) revealed at least four DOTA-avid pancreatic lesions distributed across the head, body, and tail of the pancreas, with the largest lesion located in the pancreatic head. There was no convincing evidence of DOTA-avid distant metastasis. The overall findings were suggestive of well-differentiated neuroendocrine tumor.
Abdominal MRI demonstrated a stable dominant pancreatic head lesion consistent with the known neuroendocrine tumor, with no definite nodal or hepatic metastases. Assessment of the remaining pancreatic lesions was suboptimal due to motion artifact.
Management and Hospital Course
The case was discussed at a multidisciplinary team (MDT) meeting, and the patient was admitted under the hepatobiliary surgery team for operative management. The surgical plan included a Whipple procedure (pancreaticoduodenectomy) for the dominant pancreatic head lesion with enucleation of additional pancreatic lesions. Preoperative management included close glucose monitoring and medical therapy with diazoxide and octreotide under endocrinology guidance, with continuation of lamotrigine as advised by neurology.
Surgery was performed approximately 10 months after the initial epilepsy diagnosis. Specimens submitted included peripancreatic and common hepatic artery lymph nodes, the pancreatic head mass, resection margins, additional pancreatic lesions, the body and tail of the pancreas, and the gallbladder.
Postoperative Course
Postoperatively, endocrinology recommended discontinuation of diazoxide, avoidance of dextrose infusion in the absence of hypoglycemia, continuation of octreotide 100 mcg every 8 hours, and blood glucose monitoring every 2 hours. By postoperative day 1, glucose levels had stabilized (range 6.0–10.9 mmol/L), and octreotide was subsequently discontinued. The patient experienced no further hypoglycemic episodes.
Final Histopathology
The final surgical pathology report (received approximately two weeks postoperatively) revealed the following findings: peripancreatic and common hepatic artery lymph nodes were reactive with no evidence of malignancy. The pancreatic head mass, resection margin, and additional pancreatic lesions demonstrated multiple pancreatic neuroendocrine tumors of variable sizes, with frozen section diagnoses confirmed. The body and tail of the pancreas showed multiple pancreatic neuroendocrine microadenomatosis. The gallbladder was unremarkable. These findings, taken together, established the final diagnosis of insulinomatosis.
Discussion
Insulinomatosis is a recently recognized and exceedingly rare cause of endogenous hyperinsulinemic hypoglycemia (EHH). The term was first introduced by Anlauf et al. in 2009, who compared 14 patients with multicentric insulinoma disease against 267 patients with sporadic or familial insulinomas [5]. Unlike solitary sporadic insulinoma, which accounts for approximately 90% of EHH cases and is typically curable with surgical excision, insulinomatosis is characterized by the synchronous or metachronous occurrence of multiple insulin-expressing pancreatic neuroendocrine tumors (macro- and microtumors) along with insulin-expressing monohormonal endocrine cell clusters (IMECCs), small proliferative precursor lesions less than 1 mm in size [5,6]. Critically, insulinomatosis is not associated with known hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN1), von Hippel– Lindau disease (VHL), or neurofibromatosis type 1 (NF1) [5,11] . To date, approximately 108 cases of insulinomatosis have been reported in the literature, with a marked female predominance (81%) and a mean age at diagnosis of approximately 42 years [11,16]. Our patient, a 32-year-old woman, is consistent with this demographic profile.
A particularly notable aspect of our case is the initial misdiagnosis of epilepsy, which delayed the identification of the underlying hypoglycemic etiology by approximately one year. This diagnostic pitfall is well recognized in the insulinoma literature. In a series of 59 patients with insulinoma, neurological disorders were the initial misdiagnosis in 64% of cases, with epilepsy specifically diagnosed in 39%, and 12% of patients were treated with antiepileptic drugs before the correct diagnosis was established [9]. Neuroglycopenic symptoms of hypoglycemia—including confusion, behavioral changes, seizures, and loss of consciousness—closely mimic the semiology of complex partial seizures, particularly those of temporal or frontal lobe origin [17–19] . This overlap is further compounded by several factors: insulinomas secrete insulin in a pulsatile fashion, meaning that blood glucose levels may be normal between episodes; hypoglycemia itself can trigger electroencephalographic (EEG) abnormalities that may be misinterpreted as epileptiform activity; and the paroxysmal, stereotyped nature of neuroglycopenic episodes can closely resemble seizure semiology [17,20]. In our patient, episodic hypermotor activity with facial twitching, inappropriate laughing and crying, and frontal lobe EEG activity led to a reasonable initial diagnosis of frontal lobe epilepsy. The critical clue came when a seizure episode was associated with documented hypoglycemia, prompting continuous glucose monitoring that revealed recurrent glucose values of 40–50 mg/dL. This case reinforces the importance of checking blood glucose during any seizure episode, particularly in non-diabetic patients presenting with atypical or medication-refractory seizures [18,19,21]
The biochemical evaluation of our patient followed established diagnostic protocols for EHH. The supervised 72-hour fasting test remains the gold standard, with diagnostic criteria including a plasma glucose concentration less than 55 mg/dL (3.0 mmol/L), insulin level ≥3 µU/mL (≥18 pmol/L), Cpeptide level ≥0.6 ng/mL (≥0.2 nmol/L), proinsulin level ≥5 pmol/L, and a negative sulfonylurea or meglitinide screen [3,8]. Our patient’s fasting test results demonstrated a plasma glucose of 2.6 mmol/L with inappropriately elevated insulin (66 pmol/L), Cpeptide (0.90 nmol/L [inappropriately normal relative to concurrent hypoglycemia; expected to be suppressed <0.2 nmol/L in non-insulinoma hypoglycemia]), and markedly elevated proinsulin (36.01 pmol/L, nearly five times the upper limit of normal), fulfilling all diagnostic criteria for endogenous hyperinsulinemic hypoglycemia. Insulin antibodies and oral antidiabetic drug screening were negative, effectively excluding exogenous insulin administration, autoimmune insulin syndrome (Hirata’s disease), and surreptitious sulfonylurea use [8]. Additionally, a short Synacthen test excluded adrenal insufficiency as a contributing etiology, and normal IGF-1, TSH, and FSH levels ruled out other endocrine causes of hypoglycemia.
Imaging played a pivotal role in characterizing the extent of disease in our patient. Contrast-enhanced CT initially suggested a single heterogeneously enhancing lesion in the pancreatic head, while EUS confirmed a well-demarcated 30 × 20 mm hypoechoic mass in the pancreatic head with cystic degeneration. However, the true multifocal nature of the disease was only revealed by Ga-68 DOTATATE PET/CT, which demonstrated at least four DOTA-avid lesions distributed across the head, body, and tail of the pancreas. This finding is particularly instructive, as conventional anatomic imaging (CT and MRI) has limited sensitivity for detecting small pancreatic neuroendocrine tumors, whereas Ga-68 DOTATATE PET/CT offers substantially higher localization rates, as discussed in the Introduction [14,15]. In the context of insulinomatosis, where multiple lesions of varying sizes may be present throughout the pancreas, functional imaging with somatostatin receptor–based PET/CT is particularly important for mapping disease extent and guiding surgical planning [14,22]. Our case underscores that reliance on anatomic imaging alone may underestimate the burden of disease in insulinomatosis, and that functional imaging should be strongly considered when multiple lesions are suspected.
The histopathological distinction between insulinomatosis, solitary sporadic insulinoma, and MEN1-associated insulinoma is of critical clinical importance. Solitary sporadic insulinomas are typically single, small (1–1.5 cm), benign tumors whose surgical excision results in permanent cure, with virtually no recurrence [5,7]. In contrast, MEN1-associated insulinomas are multiple but occur in the context of a germline MEN1 mutation and are associated with tumors of the parathyroid glands and anterior pituitary [4]. Insulinomatosis differs from both entities: the tumors are exclusively insulin-expressing (monohormonal), occur in the absence of MEN1 mutation, and are accompanied by the pathognomonic IMECCs proliferative insulin-positive cell clusters that are less than 1 mm in size and are believed to represent precursor lesions [5,6] . In our patient, MEN1 screening was performed given the finding of a pancreatic neuroendocrine tumor. Prolactin and chromogranin A were within normal limits. PTH was elevated at 14.75 pmol/L; however, this occurred in the context of vitamin D deficiency (25-OH vitamin D of 19.9 nmol/L) with normal adjusted calcium, a pattern most consistent with secondary hyperparathyroidism rather than primary hyperparathyroidism associated with MEN1. MEN1 genetic testing was not available during the index hospitalization but has been planned for the follow-up period; this represents a limitation of the current report. The final histopathology demonstrating multiple neuroendocrine tumors of variable sizes with microadenomatosis across the head, body, and tail of the pancreas, in the absence of MEN1-associated features— is consistent with the diagnostic criteria for insulinomatosis as defined by Anlauf et al. [5].
The molecular genetics of insulinomatosis remain incompletely understood. In 2018, Iacovazzo et al. identified a missense mutation in the MAFA gene (p. Ser64Phe, c.191C>T) as the cause of familial insulinomatosis and diabetes mellitus in two unrelated families with autosomal dominant inheritance [12]. MAFA is a β-cell–enriched transcription factor that plays a central role in regulating glucose-stimulated insulin secretion and also demonstrates oncogenic transformation potential [12]. The p. Ser64Phe mutation was shown to increase MAFA protein stability by impairing its phosphorylation-dependent degradation, leading to increased transcriptional activity and, presumably, uncontrolled β-cell proliferation [12] . A second pathogenic MAFA variant (p. Thr57Arg) was subsequently identified in a German family with a similar syndrome of insulinomatosis and mild diabetes, confirming the role of MAFA transactivation domain defects in this disease [13]. Importantly, however, no germline or somatic MAFA mutations were identified in nine patients with sporadic insulinomatosis, implying that MAFA-independent mechanisms are involved in the pathogenesis of the sporadic form [12]. Novel copy number variations involving CEBPA, FOXL2, and IRS2 have been reported in a sporadic case, suggesting potential alternative molecular pathways [6]. Our patient’s sporadic presentation, with no family history of endocrine disorders or diabetes, likely represents the more common MAFA-independent form, though genetic testing for MAFA and other candidate genes would be valuable for definitive characterization and genetic counseling.
The management of insulinomatosis presents a significant surgical challenge. Unlike solitary insulinoma, where enucleation alone is typically curative, the multicentric nature of insulinomatosis confers a substantially higher risk of persistent or recurrent hypoglycemia after conservative surgery [5,11,12]. In the original Anlauf et al. series, recurrent hypoglycemia occurred in 6 of 14 patients (43%), with a mean time to relapse of 8.4 years [5]. In the more recent Lourenço et al. series, all four patients with sporadic insulinomatosis experienced at least one recurrence, and 41% of all documented sporadic cases underwent two or more surgical resections [11]. Our patient underwent a Whipple procedure (pancreaticoduodenectomy) for the dominant pancreatic head lesion combined with resection of the body and tail, which effectively constituted a near-total pancreatectomy. This extensive approach was guided by the PET/CT findings demonstrating multifocal disease and was discussed in a multidisciplinary team (MDT) setting. Preoperative medical management with diazoxide and octreotide provided effective bridging therapy for glycemic control. Postoperatively, hypoglycemic episodes resolved promptly, and both diazoxide and octreotide were discontinued within 24 hours. The resolution of hypoglycemia is encouraging; however, it must be interpreted with caution. Notably, octreotide has been reported to achieve complete remission of a recurrent insulinomatosis lesion as a single treatment in one case, suggesting a potential role for somatostatin analogs when surgery is not feasible or when surgical eradication is incomplete [23].
This case contributes to the growing body of literature on insulinomatosis by illustrating several important clinical lessons. First, it highlights the potential for prolonged diagnostic delay when neuroglycopenic hypoglycemia manifests as seizures, reinforcing the recommendation to measure blood glucose in all patients with atypical or drug-resistant seizure presentations. Second, it demonstrates the value of multimodal imaging, particularly Ga-68 DOTATATE PET/CT, in revealing the true extent of multifocal pancreatic disease that may be underestimated by conventional imaging. Third, it underscores the importance of thorough histopathological examination of the “macroscopically normal pancreas” in patients with multiple insulinomas, as the identification of microadenomatosis and IMECCs is essential for distinguishing insulinomatosis from other causes of multicentric disease [5,11]. Finally, given the welldocumented high recurrence rate of insulinomatosis, our patient will require long-term surveillance with periodic biochemical monitoring (fasting glucose, insulin, C-peptide, and proinsulin) and imaging followup. MAFA genetic testing has been planned and will be performed at the next follow-up appointment to allow definitive molecular characterization and inform family screening. Additional large-scale molecular studies are needed to elucidate the pathogenesis of sporadic insulinomatosis and to identify potential therapeutic targets for this rare but clinically significant disease.
Limitations
This case report has several limitations that should be acknowledged. First, MEN1 genetic testing was not performed during the index admission; although biochemical screening was reassuring, germline MEN1 mutation cannot be definitively excluded without molecular confirmation. Second, MAFA genetic testing has not yet been completed, precluding definitive molecular classification of this case as familial or sporadic insulinomatosis. Third, the MRI assessment of non-dominant pancreatic lesions was suboptimal due to motion artifact, potentially underestimating the full burden of disease. Fourth, postoperative follow-up at the time of this report is limited to the early postoperative period; given the well-documented high recurrence rate of insulinomatosis (43% in the original Anlauf series), long-term biochemical and imaging surveillance will be essential to confirm durable remission. Fifth, no postoperative EEG was performed to confirm resolution of the previously observed frontal epileptiform activity. Finally, as a single-case observational report, these findings may not be generalizable, and the conclusions drawn should be interpreted within this context.
Conclusion
This case highlights several key lessons: (1) clinicians should routinely measure blood glucose during any atypical or drug-resistant seizure episode, as neuroglycopenic hypoglycemia can faithfully mimic frontal lobe seizure semiology; (2) once endogenous hyperinsulinism is confirmed, Ga-68 DOTATATE PET/CT is indispensable for delineating multifocal disease that conventional imaging may underestimate; (3) the histopathological diagnosis of insulinomatosis mandates MEN1 exclusion and consideration of MAFA genetic testing for definitive classification; and (4) given the high recurrence rate, lifelong biochemical and imaging surveillance is essential. A multidisciplinary approach integrating endocrinology, neurology, radiology, surgery,and pathology was critical for achieving biochemical cure in this patient and serves as a reminder to look beyond the brain when seizure presentations defy conventional neurological explanations.
Declarations
Consent for Publication: Written informed consent was obtained from the patient for the publication of this case report and any accompanying clinical data. A copy of the written consent is available for review by the Editor-in-Chief of this journal upon request.
Conflicts of Interest: The authors declare no conflicts of interest.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors
Author Contributions: SA contributed to conceptualization, project administration, and writing of the discussion section. SOA and SHA prepared the case presentation. AMB wrote the introduction. BSB prepared the conclusion. MAA wrote the abstract. IAA designed the graphical abstract. RAA performed journal formatting and manuscript preparation. All authors contributed to manuscript revision and approved the final version of the manuscript.