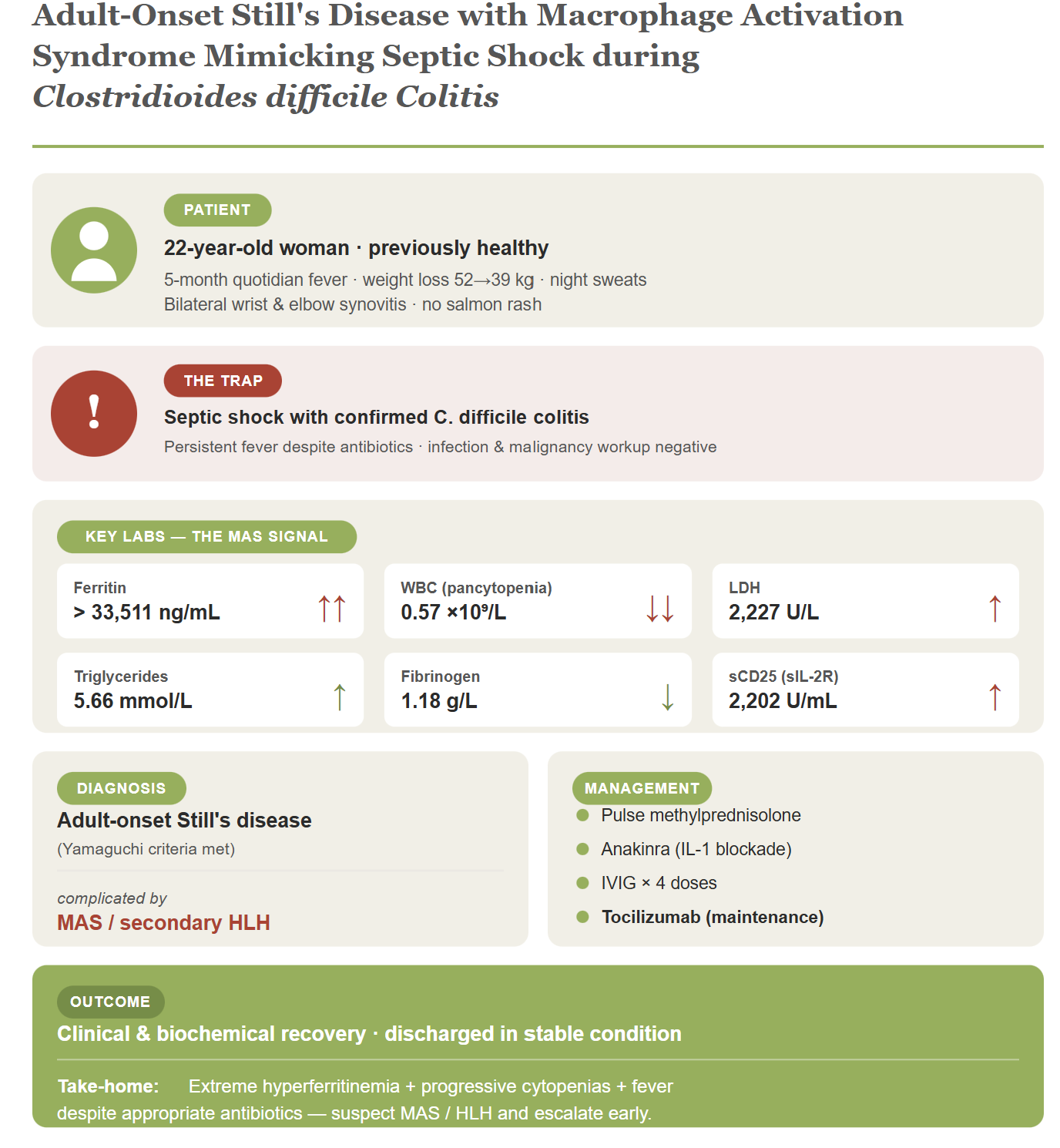
Introduction
Adult-onset Still's disease (AOSD) is an uncommon systemic autoinflammatory disorder with an estimated incidence of 0.1–0.4 per 100,000 persons per year, with a predilection for young adults and particularly women (1,2). It is classically characterized by a triad of quotidian spiking fever, polyarthritis affecting wrists, elbows, and other joints, and a transient salmon-colored rash, frequently accompanied by sore throat, lymphadenopathy, hepatosplenomegaly, and striking elevations of acute-phase reactants and serum ferritin (1–3). Despite the availability of classification criteria, AOSD remains a major diagnostic challenge because its manifestations overlap extensively with severe infections, hematologic malignancies, and other systemic inflammatory diseases, often leading to delays in diagnosis and initiation of appropriate immunomodulatory therapy (1,4,5).
A subset of patients with AOSD develop macrophage activation syndrome (MAS), a form of secondary hemophagocytic lymphohistiocytosis (HLH) that represents one of the most devastating complications of the disease (7–9). The clinical and laboratory picture of MAS frequently mimics severe sepsis, particularly when patients present with shock, organ dysfunction, and markedly elevated inflammatory markers (7–9). This overlap is especially treacherous when a true infection is concurrently documented, as prioritizing sepsis management may dangerously delay the high-dose corticosteroids and cytokine-targeted biologic agents that are essential for reversing the hyperinflammatory cascade (8,9). In this report, we describe a previously healthy young woman who presented with months-long fever of unknown origin, extreme hyperferritinemia, septic shock in the setting of documented Clostridioides difficile colitis, and progressive cytopenias, and was eventually diagnosed with AOSD complicated by MAS/secondary HLH. The case exemplifies the diagnostic pitfalls encountered when life-threatening hyperinflammation coexists with severe infection, and illustrates how timely escalation to high-dose glucocorticoids, IL-1 blockade, and adjunctive immunomodulation can lead to a favorable outcome.
Case Presentation
History of Present Illness
A 22-year-old female with no significant past medical or surgical history presented to the outpatient department for follow-up 10 days post-discharge from a clinical teaching unit (CTU) where she had been evaluated for fever of unknown origin. During the visit, she was found to be febrile and hypotensive, prompting rapid referral to the emergency department.
Her illness course began 5 months prior with constitutional symptoms including daily quotidian fever peaking mainly at night and in the late afternoon, significant unintentional weight loss from 52 kg to 39 kg (13 kg loss), night sweats, poor appetite, and profound malaise. She had been evaluated at multiple hospitals, the last of which was Aseer Hospital where she was admitted for two weeks and discharged against medical advice without an established diagnosis. Her current presentation was complicated by a 3-day history of watery, foul-smelling, non-bloody diarrhea occurring more than three times per day, and generalized abdominal pain. Review of systems showed persistent nausea and vomiting with febrile spikes, and bilateral inflammatory pain in the wrists and elbows. She mentioned a distant contact history with a tuberculosis-treated grandfather. There was no history of recent skin rash, travel, exposure to fresh or saltwater, contact with house pets or cattle, or substance use. Family history was unremarkable for autoimmunity or malignancy.
Summary of Previous Admission
The patient had been admitted under internal medicine on 23 July 2025 for workup of B symptoms with an initial impression of malignancy. Computed tomography (CT) revealed multiple enlarged supra- and infra-diaphragmatic lymph nodes and mild splenomegaly (13.5 cm). Positron emission tomography (PET) scan demonstrated consistent findings along with diffuse bone marrow uptake. A right inguinal lymph node biopsy on 27 July 2025 showed reactive lymph node with dermatopathic changes, no granulomata, and was negative for malignancy. A repeat axillary lymph node biopsy was taken on 3 August 2025 with a plan to follow up in clinic. During this admission, ferritin was 12,224.0 ng/mL, LDH was elevated (880 U/L initially, declining to 514 U/L on follow-up), and AST trended upward from 136 to a peak of 629 U/L. Platelet count ranged from 318,000 to 547,000/cmm and hemoglobin ranged from 78 to 95 g/L.
An extensive infectious workup during this admission was negative, including blood cultures, urine cultures, tissue culture, hepatitis panel, HIV, syphilis, CMV/EBV serologies, malaria, QuantiFERON-TB Gold/TB PCR, Brucella, Q fever, Parvovirus, Dengue, and Toxoplasmosis. Rheumatology workup was negative for ANA, anti-dsDNA, RF, anti-CCP, ANCA, and cryoglobulins; complement levels were within normal limits. Rheumatology's initial impression favored lymphoproliferative disorder or malignancy over connective tissue disease. The patient was discharged while still febrile and tachycardic to continue follow-up as outpatient.
Physical Examination
On examination, the patient appeared acutely ill (GCS 15/15), lethargic, and cachectic with a current weight of 39 kg, with signs of severe dehydration including dry mucous membranes and delayed capillary refill of more than two seconds. She was hemodynamically unstable with hypotension (SBP 73–93 mmHg, DBP 42–55 mmHg), tachycardia (heart rate 131–141 bpm), and fever (38.4–38.9°C), requiring intensive care admission for inotropic support.
Head and neck examination revealed no palpable lymphadenopathy. Musculoskeletal assessment revealed active synovitis of the bilateral wrists and metacarpophalangeal (MCP) joints, characterized by palpable swelling and tenderness. Skin examination was significant for bilateral hyperpigmentation over the upper extremities and left thigh; the classic evanescent salmon-colored rash was absent. Abdominal examination demonstrated a soft lax abdomen with diffuse generalized tenderness and no evidence of organomegaly on palpation. Cardiovascular examination revealed normal S1 and S2 with no murmurs. Respiratory examination showed equal bilateral air entry with no added sounds. Neurological examination was unremarkable with normal tone, power 5/5 bilaterally, intact sensation, and normal reflexes. There was no lower limb edema or signs of deep vein thrombosis.
Key Investigations
The initial hematological laboratory investigations for the current presentation were remarkable for severe microcytic hypochromic anemia demonstrated by 8.3 g/dL hemoglobin, mean corpuscular volume (MCV) of 68.2 fL, and a mean corpuscular hemoglobin (MCH) of 22.6 pg. Additionally, there was marked leukocytosis (20.9 × 10⁹/L) with neutrophilia measured as 18.81 × 10⁹/L and lymphopenia at 0.84 × 10⁹/L (Table 1a). Inflammatory markers showed extreme hyperferritinemia (17,347.7 ng/mL), an elevated C-reactive protein (CRP) of 104.80 mg/L, erythrocyte sedimentation rate (ESR) of 64 mm/hr, and procalcitonin (PCT) of 0.78 ng/mL (Table 1b).
Table 1a. Hematology Results
Parameter | Patient Value | Reference Range |
Hemoglobin | 8.3 g/dL | 12–16 g/dL |
Hematocrit | 24.3% | 36–46% |
RBC | 3.56 ×10⁶/mm³ | 3.5–6.0 ×10⁶/mm³ |
MCV | 68.2 fL | 80–100 fL |
MCH | 22.6 pg | 27–32 pg |
MCHC | 33.1 g/dL | 30–35 g/dL |
RDW | 20.6% | 11.5–14.5% |
Total Leukocytic Count | 20,900/cmm | 4,000–11,000/cmm |
Neutrophils | 18,810 cells/mm³ | 2,000–7,500 cells/mm³ |
Lymphocytes | 840 cells/mm³ | 1,500–4,500 cells/mm³ |
Monocytes | 630 cells/mm³ | 200–800 cells/mm³ |
Eosinophils | 210 cells/mm³ | 40–400 cells/mm³ |
Platelets | 495,000/cmm | 150,000–450,000/cmm |
Table 1b. Biochemical and Inflammatory Markers
Parameter | Patient Value | Reference Range |
Ferritin | 17,347.7 ng/mL | 20–250 ng/mL |
CRP | 104.80 mg/L | <5.0 mg/L |
ESR | 64 mm/hr | 0–20 mm/hr |
PCT | 0.78 ng/mL | <0.5 ng/mL |
Iron | 1.70 µmol/L | 11–30 µmol/L |
TIBC | 18.6 µmol/L | 45–81 µmol/L |
LDH | 880 U/L | 125–220 U/L |
Total Bilirubin | 5.2 µmol/L | <17 µmol/L |
AST | 46 U/L | <40 U/L |
ALT | 14 U/L | <41 U/L |
ALP | 119 U/L | 40–130 U/L |
Albumin | 31 g/L | 35–50 g/L |
Total Protein | 65 g/L | 60–80 g/L |
INR | 1.13 | 0.8–1.2 |
PT | 13.00 s | 11–13.5 s |
PTT | 27.90 s | 25–35 s |
The arterial blood gas revealed respiratory alkalosis with pH 7.49, PCO₂ 29 mmHg, and relatively normal bicarbonate (HCO₃) of 22.1 mmol/L. Oxygenation markers showed a PaO₂ of 61 mmHg, oxygen saturation of 91.3%, and O₂Hb of 88.4%. The renal and electrolyte profile showed mild electrolyte imbalances, specifically hyponatremia and borderline hypokalemia (Table 2).
Table 2. Kidney Function and Electrolyte Profile
Parameter | Patient Value | Reference Range |
Creatinine | 44 µmol/L | 60–110 µmol/L |
BUN | 2.7 mmol/L | 2.5–7.1 mmol/L |
eGFR | 166 mL/min | >90 mL/min |
Sodium | 133 mmol/L | 135–145 mmol/L |
Potassium | 3.5 mmol/L | 3.5–5.2 mmol/L |
Chloride | 103 mmol/L | 98–107 mmol/L |
Calcium | 2.09 mmol/L | 2.12–2.62 mmol/L |
Magnesium | 0.73 mmol/L | 0.85–1.10 mmol/L |
AGAP | 16 mEq/L | 6–12 mEq/L |
Uric Acid | 269 µmol/L | 155–357 µmol/L |
Phosphorus | 1.03 mmol/L | 0.81–1.45 mmol/L |
A comprehensive multiplex PCR stool panel tested positive for Clostridioides difficile toxin A/B, Enteroaggregative E. coli (EAEC), and Enteropathogenic E. coli (EPEC), while ruling out a wide array of other gastrointestinal pathogens including Campylobacter, Salmonella, Shigella, Vibrio, Cryptosporidium, Giardia, and numerous enteric viruses (Norovirus, Rotavirus, Adenovirus, Astrovirus, Sapovirus). A comprehensive respiratory panel was completely negative for all tested viral and bacterial targets, including SARS-CoV-2, Influenza A/B, RSV, Parainfluenza 1–4, Mycoplasma pneumoniae, Bordetella pertussis, Chlamydophila pneumoniae, and MERS-CoV. All blood cultures drawn as part of the septic screen remained persistently negative throughout the hospital course.
Immunological workup, including antinuclear antibody (ANA), anti-dsDNA, rheumatoid factor (RF), anti-CCP, anti-SS-A, anti-SS-B, MPO-ANCA, PR3-ANCA, and cryoglobulins, was negative. Notably, the repeated anti-dsDNA (ELISA) returned a value of 51.56, which was below the positive threshold but in the borderline range, compared with a clearly negative result on the initial admission. Complement C3 was within normal limits (0.735 g/L); however, C4 was low at 0.06 g/L (reference 0.10–0.40 g/L), which may reflect complement consumption in the setting of systemic inflammation. Despite the borderline anti-dsDNA value and the low C4, systemic lupus erythematosus (SLE) was not pursued further because the patient did not meet any other 2019 EULAR/ACR SLE classification criteria: there was no mucocutaneous, renal, serosal, hematologic (autoimmune), or neuropsychiatric involvement attributable to SLE; ANA was negative; and the clinical phenotype was dominated by quotidian fever, polyarthritis of the wrists and MCPs, extreme hyperferritinemia, and progression to MAS, which is far more consistent with AOSD. The isolated low C4 was interpreted as complement consumption in the context of systemic hyperinflammation rather than evidence of an underlying connective tissue disease. These findings further supported the clinical diagnosis by excluding connective tissue diseases.
Imaging
A portable anteroposterior thoracic radiograph revealed diffuse, bilateral interstitial opacities and prominent reticular markings throughout both lung fields, without obvious large focal consolidations, large pleural effusions, or pneumothoraces. The cardiomediastinal silhouette appeared generally within normal limits. Multiple external ECG leads and wires were seen overlying the patient's chest. ECG demonstrated sinus tachycardia. CT abdomen was unremarkable for ileus or megacolon.
Hospital Course and Management
The patient was admitted to the ICU for inotropic support and started on empirical broad-spectrum antibiotics, alongside oral vancomycin and IV metronidazole for fulminant C. difficile colitis. CT abdomen was unremarkable for ileus or megacolon. She was shifted back to the floor after two days of hemodynamic stabilization. Hematology was consulted and the patient was scheduled for bone marrow biopsy. She continued to spike high-grade fevers throughout.
Gastroenterology was consulted for persistent watery diarrhea and hyperpigmentation; upper endoscopy (EGD) was performed to rule out Whipple's disease, with unremarkable scope findings and biopsy. The oral vancomycin course was extended to 14 days for ongoing diarrhea. During this period, the patient developed features of preseptal cellulitis versus dacryoadenitis, confirmed by orbital CT and ophthalmology assessment.
She subsequently became hypotensive again, requiring a second ICU admission with an impression of sepsis/hypovolemia. She was started on inotropic support and empirical meropenem, to which she responded well. Relevant investigations during this period showed CRP 105.50 mg/L (rising from 104.80), PCT 0.17 (declining from 0.78 on initial presentation), ferritin declining to 8,692.8 ng/mL, hemoglobin 89 g/L, WBC 19.20 × 10⁹/L, platelets 549,000/cmm, AST 51 U/L, ALT 8 U/L, ALP 224 U/L, albumin 28 g/L, and INR 1.19.
Bone marrow biopsy from the unilateral posterior iliac crest (aspirate smears, clot section, trephine biopsy, and peripheral blood) returned the following: normocellular bone marrow with increased granulopoiesis, megakaryopoiesis, and normal erythropoiesis; no immunomorphological evidence of hematolymphoid malignancy in the provided sample. Hemophagocytosis was not confirmed on the reviewed bone marrow specimen.
After stabilization and discharge from the ICU on meropenem, oral vancomycin, and resumed IV metronidazole, the patient continued to spike high-grade fevers with persistently negative cultures. Rheumatology was contacted again; their impression initially favored a lymphoproliferative disorder versus infection, as ferritin was declining without immunosuppressive therapy. However, repeated laboratory workup was notable for: ferritin >33,511 ng/mL, LDH 1,010 U/L, triglycerides 5.88 mmol/L, fibrinogen 1.44 g/L (low), CRP 50.60 mg/L (rising to 102.90), PCT 0.48 (rising to 3.25), ESR 33 mm/hr, AST 107 U/L, INR 1.22, and PT 13.50 seconds.
She was subsequently transferred to rheumatology's care as a case of Still's disease and was started on methylprednisolone 100 mg BID. Repeated labs two days afterward showed dramatic worsening with pancytopenia: WBC crashed to 0.57 × 10⁹/L (neutrophils 0.23 × 10⁹/L, lymphocytes 0.24 × 10⁹/L), hemoglobin 78 g/L, platelets 199,000/cmm, ferritin >33,511 ng/mL, LDH surging to 2,227 U/L, AST 318 U/L (rising from 181 U/L), ALT 126 U/L (improving to 17), triglycerides 5.66 mmol/L, fibrinogen 1.18 g/L, and soluble IL-2 receptor (sCD25) 2,202 U/mL. These findings were consistent with the progression to macrophage activation syndrome (MAS)/secondary HLH. The evolution of ferritin, LDH, WBC, and platelet count across the clinical course is summarized in Figure 1.
Hematology consultation concluded that AOSD was progressing into MAS/HLH. The patient was escalated to pulse-dose methylprednisolone, anakinra, and 4 doses of intravenous immunoglobulin (IVIG). The diagnosis of AOSD was established by fulfilment of the Yamaguchi criteria (11): three major criteria quotidian fever >39°C for more than one week, arthralgia/arthritis lasting more than two weeks (bilateral wrist and MCP synovitis), and leukocytosis >10,000/mm³ with >80% granulocytes (WBC 20,900 with 90% neutrophils) and two minor criteria lymphadenopathy and/or splenomegaly (radiologically confirmed) and a negative ANA and RF. The classical evanescent rash, which constitutes the fourth major criterion, was absent.
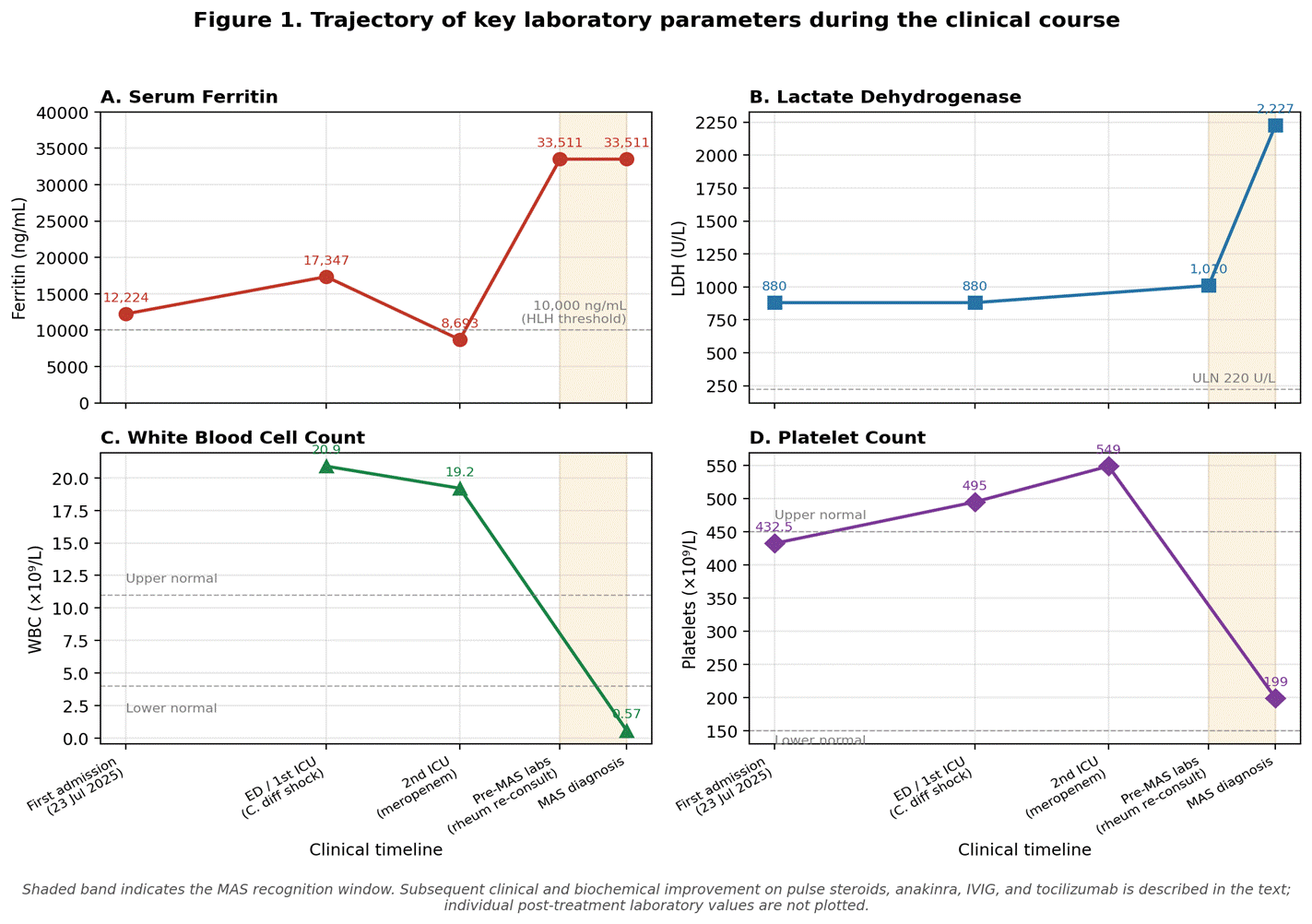
Figure 1. Trajectory of key laboratory parameters during the clinical course. (A) Serum ferritin, (B) lactate dehydrogenase (LDH), (C) white blood cell count, and (D) platelet count at successive clinical milestones, plotted using values reported in the case narrative. The shaded band indicates the recognition window for MAS. Subsequent improvement on pulse-dose corticosteroids, anakinra, IVIG, and tocilizumab was clinical and biochemical; individual post-treatment laboratory values are not plotted.
Follow-up
On the targeted regimen, she demonstrated gradual improvement over one month. She was discharged from the hospital in stable condition and transitioned to tocilizumab therapy. Her outpatient care plan involves continued follow-up with the rheumatology department to manage her condition and monitor her ongoing response to treatment.
Timeline
Date | Clinical Status and Milestones |
Early 2025 | Onset of constitutional symptoms (fever, weight loss of 52 to 39 kg, night sweats). Seen at multiple hospitals including Aseer Hospital (discharged AMA, no diagnosis). |
23 Jul 2025 | First admission (FUO workup): CT showed supra/infra-diaphragmatic lymphadenopathy and mild splenomegaly (13.5 cm); PET showed diffuse BM uptake; Ferritin 12,224 ng/mL; LDH 880 U/L. |
27 Jul 2025 | Right inguinal lymph node biopsy: Reactive with dermatopathic changes. No granuloma. Negative for malignancy. |
3 Aug 2025 | Axillary lymph node biopsy taken. Patient discharged while still febrile for OPD follow-up. |
Late Aug 2025 | ER/ICU admission: Hypotensive shock, C. difficile colitis, Ferritin 17,347 ng/mL. Started on oral vancomycin, IV metronidazole, broad-spectrum antibiotics. |
Hospital course | Second ICU admission for recurrent hypotension; meropenem started. Bone marrow biopsy: normocellular, no malignancy. EGD to rule out Whipple's: unremarkable. Ferritin declined to 8,693 then surged to >33,511 ng/mL. |
Early Sep 2025 | Transferred to Rheumatology. Diagnosis of AOSD confirmed. Labs showed pancytopenia (WBC 0.57), LDH 2,227, fibrinogen 1.18, triglycerides 5.66, sCD25 2,202 consistent with MAS/HLH. |
Mid Sep 2025 | Escalation to pulse steroids, anakinra, and 4 doses of IVIG. |
Discharge | Significant recovery over 1 month; discharged on tocilizumab. |
Discussion
Adult-onset Still's disease (AOSD) is a rare systemic autoinflammatory disorder with an estimated annual incidence of 0.1–0.4 per 100,000 (1,2). The Yamaguchi classification criteria remain the most widely used diagnostic tool, with a reported sensitivity of 96.2% and specificity of 92.1% (11). The 2024 EULAR/PReS recommendations have recognized that systemic juvenile idiopathic arthritis and AOSD represent a single disease entity now termed “Still's disease”and endorse the early use of IL-1 or IL-6 inhibitors combined with short-duration glucocorticoids as the optimal therapeutic strategy (12).
Our patient's illness evolved over five months with quotidian fevers, 13 kg weight loss, bilateral wrist and elbow synovitis, marked leukocytosis with neutrophilia, and extreme hyperferritinemia that escalated from 12,224 ng/mL to 17,347 ng/mL and ultimately to >33,511 ng/mL, fulfilling the Yamaguchi criteria (three major and two minor) (11). The absence of the classic salmon-colored rash is consistent with reports that the rash may be absent in a substantial proportion of AOSD patients without precluding the diagnosis (3,5). The exhaustive negative infectious and malignancy workups including negative cultures, viral serologies, two reactive lymph node biopsies, and a bone marrow biopsy showing no malignancy were crucial in establishing AOSD as a diagnosis of exclusion (13).
Markedly elevated serum ferritin exceeding five times the upper limit of normal is a hallmark of AOSD, with a reported sensitivity of approximately 80% and specificity of 41% when used alone; combining this with glycosylated ferritin ≤20% increases specificity to 93% (14). Ferritin levels exceeding 10,000 ng/mL demonstrate 90% sensitivity and 96% specificity for HLH (15). In our case, the ferritin trajectory from 12,224 to >33,511 ng/mL served as both a diagnostic and prognostic marker, heralding the evolution into MAS.
MAS, occurring in approximately 12–17% of AOSD patients with a mortality exceeding 20% (2,7), was diagnosed in our patient based on the dramatic development of pancytopenia (WBC 0.57 × 10⁹/L, Hgb 78 g/L, platelets 199,000/cmm), hyperferritinemia >33,511 ng/mL, elevated LDH (2,227 U/L), hypertriglyceridemia (5.66 mmol/L), hypofibrinogenemia (1.18 g/L), elevated sCD25 (2,202 U/mL), and hepatic dysfunction (AST 318 U/L). These findings are consistent with the 2022 EULAR/ACR points to consider for HLH/MAS, which emphasize the importance of monitoring the evolution of laboratory parameters over time rather than relying on any single abnormality (16). A critically challenging feature of this case was the concurrent C. difficile colitis with septic shock. The overlap between MAS/HLH and severe sepsis poses one of the most difficult diagnostic dilemmas in critical care, as both conditions share features of fever, organ dysfunction, and elevated inflammatory markers (17). However, extreme hyperferritinemia (>10,000 ng/mL), progressive multilineage cytopenias, and persistence of high fevers despite appropriate antimicrobial therapy are features that favor MAS/HLH over isolated sepsis (17). Importantly, infections can serve as triggers for MAS in patients with underlying autoinflammatory conditions, and the two processes may coexist (7,18). In our patient, C. difficile colitis likely served as a secondary trigger that precipitated fulminant MAS in the setting of already dysregulated immunity. Management followed a stepwise escalation aligned with current recommendations. After initial stabilization with antibiotics and targeted C. difficile treatment, recognition of AOSD led to methylprednisolone. The development of MAS prompted escalation to pulse steroids, anakinra, and IVIG. The 2024 EULAR/PReS recommendations endorse high-dose glucocorticoids as the foundation of MAS therapy, with anakinra, cyclosporine, and/or interferon-γ inhibitors as part of initial therapy (12). Meta-analytic data demonstrate complete remission rates of approximately 73% with anakinra and 80% with tocilizumab in AOSD (19). Following acute stabilization, the transition to tocilizumab for maintenance therapy represents a rational strategy addressing both the immediate cytokine storm and long-term disease control (12,19).
Limitations
This report has several limitations. First, it describes a single patient, and therefore the diagnostic and therapeutic conclusions cannot be generalized. Second, hemophagocytosis was not confirmed on the reviewed bone marrow specimen; while this does not preclude the diagnosis of MAS/secondary HLH given the otherwise classical clinical and laboratory profile, its absence weakens histopathological confirmation. Third, the HScore could not be reliably reconstructed retrospectively, as the maximum recorded temperature during the MAS phase and other component variables required for the score were not all systematically documented at the same time point. Fourth, follow-up under tocilizumab maintenance therapy was limited to the immediate post-discharge period, and longer-term outcomes regarding sustained remission, relapse, and steroid tapering are not yet available. Finally, the temporal association between Clostridioides difficile colitis and the onset of fulminant MAS is suggestive but does not establish causation; whether the infection acted as a true trigger or as a coincident process cannot be determined from a single case.
Clinical Pearls
- In a febrile young adult with negative infectious and malignancy workup, extreme hyperferritinemia (>10,000 ng/mL) should immediately raise suspicion for AOSD and its hyperinflammatory complication, MAS/secondary HLH.
- Persistent high-grade fever and rising inflammatory markers despite appropriate, source-controlled antimicrobial therapy in a patient with documented infection should prompt re-evaluation for an underlying hyperinflammatory syndrome rather than escalation of antibiotics alone.
- Trends matter more than isolated values: a falling ferritin or platelet count from a markedly abnormal baseline can paradoxically herald progression to MAS rather than recovery.
- Concurrent infection does not exclude MAS; infections frequently act as triggers in patients with underlying autoinflammatory predisposition, and the two processes can require simultaneous management.
- Early multidisciplinary collaboration (rheumatology, hematology, critical care, and infectious diseases) and timely escalation to high-dose glucocorticoids, IL-1 blockade, and adjunctive immunomodulation are essential for survival.
Conclusion
This case underscores the diagnostic and therapeutic challenges of adult-onset Still's disease complicated by macrophage activation syndrome, particularly when it coexists with severe infection such as Clostridioides difficile colitis. The significant overlap between MAS/HLH and septic shock can delay recognition of the underlying hyperinflammatory state, with potentially life-threatening consequences. In our patient, extreme hyperferritinemia, progressive cytopenias, and persistent fever despite appropriate antimicrobial therapy were key features that guided the diagnosis.
Importantly, this case highlights the need for a high index of suspicion for MAS in patients with unexplained systemic inflammation and rapidly evolving laboratory abnormalities. The prompt escalation to high-dose corticosteroids, IL-1 blockade, and adjunctive immunomodulatory therapy resulted in a favorable outcome, emphasizing the importance of early and aggressive treatment.
Clinicians should remain vigilant for the coexistence of infection and hyperinflammatory syndromes, recognizing that infections may act as triggers rather than sole explanations for clinical deterioration. Early multidisciplinary collaboration and timely initiation of targeted therapy are essential to improving survival in this complex and potentially fatal condition.
Declarations
Conflicts of Interest: The authors declare no conflicts of interest.
Funding: None.
Acknowledgements: None.
Ethical Approval: Not required for case reports in accordance with institutional policy.
Consent for Publication: Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Author Contributions: All authors contributed to the conception, drafting, and revision of the manuscript and approved the final version.
Data Availability: Data sharing is not applicable as no datasets were generated or analyzed.